PennCNV individual-based CNV calling
Suppose we already generated the necessary input signal intensity file, including sample1.txt, sample2.txt and sample3.txt, as described in the previous tutorial section in Input Files. Below is a description of the procedure for CNV calling for each of the three individuals using PennCNV.
1 General CNV calling overview
2 CNV calling on three individuals
3 GCmodel adjustment
General CNV calling overview
A general flowchart for CNV calling algorithm by PennCNV is described in the figure below:
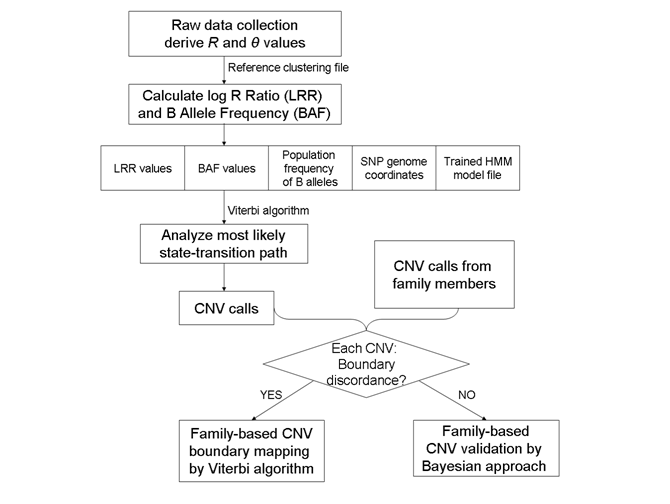
As we can see from the flowchart, for CNV calling, we need to have LRR and BAF values (contained within the signal intensity file sample*.txt), the population frequency of B alleles and SNP genome coordinates (contained within the file hh550.hg18.pfb), and an appropriate HMM model (contained within the hh550.hmm file).
Notes: the hh550.hg17.pfb and hh550.hg18.pfb file contains the genome coordinates and the population frequency for each SNP in the HumanHap550 array. The differences are that they used different genome assembly: hg17 corresponds to the 2004 human genome assembly (or NCBI35 assembly), while the hg18 corresponds to the 2006 human genome assembly (or NCBI 36 assembly). In the latest release of PennCNV, the hg17 files have been removed from the package.
CNV calling on three individuals
Next we can start calling CNVs for these three individuals. Suppose we are already inside the penncnv/ directory:
[kai@adenine penncnv]$ detect_cnv.pl -test -hmm lib/hh550.hmm -pfb lib/hh550.hg18.pfb sample*.txt -log sampleall.log -out sampleall.rawcnv
If you are using Windows, or if the above command does not work and show “bash: detect_cnv.pl: command not found” error message, try to add “perl” in the beginning of the command line before “detect_cnv.pl”.
If you are using Windows shell, the “sample*.txt” won’t be recognized as the expansion to 3 files. Try to use “sample1.txt sample2.txt sample3.txt” explicitly in command line, or you can put the three file names into a list file (see tips below) and use the --list argument.
In the command line, we have several arguments: the --test argument tells the program to generate CNV calls, the --hmmfile and --pfbfile arguments tell the program to use a HMM model file called lib/hh550.hmm and a Population Frequency of B Allele file called hh550.hg18.pfb. The sample*.txt notation tells the program to generate CNV calls for three files (sample1.txt, sample2.txt and sample3.txt), the –log argument tells the program to write log information to the sampleall.log file, while the –out argument tells the program to output CNV calls to a file called sampleall.rawcnv. This should be done in a few minutes in a modern computer.
Tip: Instead of listing all file names in the command line, we can generate a listfile containing file names to be processed (one per line), then use the --list argument in the command line. This is especially useful when you want to process a subset of all files in a directory, or when you have too many (>2000) files in a directory (in this case, the expansion of sample*.txt exceed the maximum length of allowable characters in a command line).
Note: By default only autosome CNVs will be detected, but the --chrx argument can be used in the above command to generate CNV calls for chromosome X only. The CNV calling for chrX is slightly different from that of autosomes. It is highly recommended to use the --sexfile argument to supply gender annotation for all genotyped samples. The sexfile is two-column file, with first column as signal file name, and second column as either male or female. If sex for a sample is not provided in sexfile, or if --sexfile is not specified, PennCNV will try to predict the gender of the sample, based on BAF heterozgous rate (fraction of chrX markers with BAF between 0.25 and 0.75) of >0.1 (female) or <0.1 (male). Next, PennCNV will adjust the LRR values by a fixed value such that females have median LRR at 0 and males have median LRR at the same value as that for CN=1 in the HMM file. After this step, the CNV calling is then applied in a similar way as autosomes.
We can then take a look at the resulting sampleall.rawcnv file. The first a few lines look like this:
chr1:156783977-156788016 numsnp=6 length=4,040 state2,cn=1 sample1.txt startsnp=rs16840314 endsnp=rs10489835
chr2:4191253-4200019 numsnp=3 length=8,767 state2,cn=1 sample1.txt startsnp=rs1175867 endsnp=rs1175854
chr2:183794033-183797494 numsnp=3 length=3,462 state2,cn=1 sample1.txt startsnp=rs17758247 endsnp=rs1462530
chr2:242565979-242656041 numsnp=16 length=90,063 state2,cn=1 sample1.txt startsnp=rs12987376 endsnp=rs6740738
chr5:17671545-17780897 numsnp=15 length=109,353 state2,cn=1 sample1.txt startsnp=rs6894435 endsnp=rs6451161
chr10:58185524-58189264 numsnp=6 length=3,741 state2,cn=1 sample1.txt startsnp=rs12250344 endsnp=rs11005504
chr11:55127597-55204003 numsnp=11 length=76,407 state2,cn=1 sample1.txt startsnp=rs2456022 endsnp=rs7934845
chr19:503650-507632 numsnp=4 length=3,983 state2,cn=1 sample1.txt startsnp=rs7247601 endsnp=rs4919850
chr1:59077355-59078584 numsnp=3 length=1,230 state5,cn=3 sample1.txt startsnp=rs942123 endsnp=rs3015321
chr1:147305744-147427061 numsnp=7 length=121,318 state5,cn=3 sample1.txt startsnp=rs11579261 endsnp=rs3853524
The first field in each line specifies the chromosome region. Be careful here! Although the sample*.txt file also contains chromosome coordinates for each SNP, this information is ignored during CNV calling. Instead, the chromosome coordinates for each SNP is read from the PFB file (hh550.hg18.pfb), which was generated using the chromosome coordinates for the 2006 human genome assembly, so the chromosome regions shown in the sampleall.rawcnv file are for 2006 human genome assembly. If we want to generate CNV calls for the 2004 human genome assembly, we will need to use a different PFB file (hh550.hg17.pfb). This design allows us to easily switch CNV calls for different genome assembly without changing the signal intensity files. This also means that the input signal intensity file does not have to contain the Chr and Position column: as they will be ignored by default. However, if you insist to use the genome coordinates annotation in your own input file (for whatever reasons), you can use the --coordinate_from_input argument, which will apply the Chr and Position annotation in input files, rather than PFB files.
The second field and the third field in each line in sampleall.rawcnv tell how many SNPs are contained within the CNV and the length of the CNV (calculated by subtracting the coordinate of the last SNP by the coordinate of the first SNP plus 1).
The fourth field tells the HMM state and the actual copy number (CN) of the CNV call. You can ignore the state, as it is jus an internal HMM parameter. The CN refers to the actual integer copy number estimates, and the normal copy number is 2. So for autosome, CN=0 or 1 means there is a deletion and CN>=3 means there is a duplication. For chrX or chrY in males, CN=1 is the normal copy number and CN=0 means a deletion.
The fifth, sixth and seventh fields specify the original file name, the starting marker identifier and the ending marker identifier in the CNV, respectively.
There are a total of 51 CNVs generated in this step for three individuals. Of the 17 CNVs in offspring, 12 are inherited from parents, and 5 are Not Detected in Parents (so-called CNV-NDPs). These CNV-NDPs may be false positive calls in offspring, or false negative calls in parents, or as de novo CNVs. Of the 12 inherited CNVs, 10 have identical boundaries as the corresponding CNVs in father or mother. Of the 5 CNV-NDPs, four of them contain less than 10 SNPs, but one of them is quite large (50 SNPs) and does not overlap with any immunoglobulin region.
We can then take a look at the sampleall.log file, which contains information reported by the program during analysis of samples:
[kai@beta penncnv]$ cat sampleall.log
NOTICE: Reading marker coordinates and population frequency of B allele (PFB) from penncnv/lib/hh550.hg18.pfb ... Done with 561288 records (178 records in chr M,XY were discarded)
NOTICE: Reading LRR and BAF for chr1-22 from sample1.txt ... Done with 561288 records in 24 chromosomes (178 records are discarded due to lack of PFB information)
NOTICE: Data from chromosome X,Y will not be used in analysis
NOTICE: Median-adjusting BAF values for all autosome markers from sample1.txt by 0.0328
NOTICE: quality summary for sample1.txt: LRR_mean=0.0038 LRR_median=0.0000 LRR_SD=0.1383 BAF_mean=0.5046 BAF_median=0.5000 BAF_SD=0.0425 BAF_DRIFT=0.000151 WF=0.0120 GCWF=0.0034
NOTICE: Reading LRR and BAF for chr1-22 from sample2.txt ... Done with 561288 records in 24 chromosomes (178 records are discarded due to lack of PFB information)
NOTICE: Data from chromosome X,Y will not be used in analysis
NOTICE: Median-adjusting BAF values for all autosome markers from sample2.txt by 0.0031
NOTICE: quality summary for sample2.txt: LRR_mean=0.0034 LRR_median=0.0000 LRR_SD=0.1338 BAF_mean=0.5063 BAF_median=0.5000 BAF_SD=0.0391 BAF_DRIFT=0.000070 WF=0.0194 GCWF=0.0144
NOTICE: Reading LRR and BAF for chr1-22 from sample3.txt ... Done with 561288 records in 24 chromosomes (178 records are discarded due to lack of PFB information)
NOTICE: Data from chromosome X,Y will not be used in analysis
NOTICE: Median-adjusting BAF values for all autosome markers from sample3.txt by 0.0268
NOTICE: quality summary for sample3.txt: LRR_mean=0.0022 LRR_median=0.0000 LRR_SD=0.1260 BAF_mean=0.5045 BAF_median=0.5000 BAF_SD=0.0431 BAF_DRIFT=0.000186 WF=-0.0177 GCWF=-0.0104
Note: As of June 2008, the –medianadjust argument is turned on by default in the program to reduce false positive duplication calls for problematic samples. The effect is that the BAF_median measure for all samples are automatically adjusted to be 0.5. If the user wants to reproduce the results from the previous version of PennCNV, you can turn the argument off by --nomedianadjust argument.
As we can see from the log message, first the program reads the PFB file, and it discarded 178 records in chr M, Y and XY. Next the program reads signal information for autosomes from the sample1.txt file, and discarded 178 records in the signal file (The reason is that these records/SNPs is not read/annotated in the PFB file). Note that a total of 561288 markers are read into PennCNV for analysis: this sounds right for Illumina 550K arrays. For Affy GW6 or GW5 array, this number should be around 1.8 million or 800K; for Illumina 1M array, this number should be around 1M. If it's substantially lower than the expected number, then it means something is wrong so that the signal files are not read correctly. Examine the signal files to see how many lines (markers) it has, and whether the last line is complete, to check the possibility of file corruption or file incompletion during generation.
Next the program prints out a list of sample quality summary: this information is very useful step in quality control (see http://www.neurogenome.org/cnv/penncnv/qc_tutorial.htm for more detail). If some of the quality measure looks bad, a warning message will be printed out that the sample does not pass quality control criteria. The CNV calls will still be made for all samples regardless of quality, but users are advised to take caution in analyzing CNV calls from low-quality samples.
GCmodel adjustment
The current version of PennCNV now implements an improved version of wave adjustment procedure for genomic waves via the –gcmodel argument. The algorithm is described in detail in the Diskin et al paper. If a large fraction of your samples have waviness factor (WF value) less than -0.04 or higher than 0.04, it is much better to apply the adjustment procedure to reduce false positive calls.
For the 3 samples in the tutorial, the quality is actually quite good so signal adjustment has little effect. Nevertheless, we can try to do the adjustment with the following command:
[kai@adenine penncnv]$ detect_cnv.pl -test -hmm lib/hh550.hmm -pfb lib/hh550.hg18.pfb sample1.txt sample2.txt sample3.txt -log sampleall.adjusted.log -out sampleall.adjusted.rawcnv -gcmodel hh550.hg18.gcmodel
This will apply the GC-model specified in hh550.hg18.gcmodel for signal adjustment, before generating CNV calls. The results will be written to sampleall.adjusted.rawcnv file. The following LOG information will be printed out:
NOTICE: All program notification/warning messages that appear in STDERR will be also written to log file sampleall.adjusted.log
NOTICE: Reading marker coordinates and population frequency of B allele (PFB) from penncnv/lib/hh550.hg18.pfb ... Done with 561288 records (178 records in chr M,XY were discarded)
NOTICE: Reading LRR and BAF for chr1-22 from sample1.txt ... Done with 561288 records in 24 chromosomes (178 records are discarded due to lack of PFB information)
NOTICE: Adjusting LRR by GC model: WF changes from 0.0120 to 0.0129, GCWF changes from 0.0034 to 0.0057
NOTICE: Data from chromosome X,Y will not be used in analysis
NOTICE: Median-adjusting BAF values for all autosome markers from sample1.txt by 0.0328
NOTICE: quality summary for sample1.txt: LRR_mean=0.0037 LRR_median=0.0000 LRR_SD=0.1384 BAF_mean=0.5046 BAF_median=0.5000 BAF_SD=0.0425 BAF_DRIFT=0.000151 WF=0.0129 GCWF=0.0057
NOTICE: Reading LRR and BAF for chr1-22 from sample2.txt ... Done with 561288 records in 24 chromosomes (178 records are discarded due to lack of PFB information)
NOTICE: Adjusting LRR by GC model: WF changes from 0.0194 to -0.0113, GCWF changes from 0.0144 to -0.0035
NOTICE: Data from chromosome X,Y will not be used in analysis
NOTICE: Median-adjusting BAF values for all autosome markers from sample2.txt by 0.0031
NOTICE: quality summary for sample2.txt: LRR_mean=0.0032 LRR_median=0.0000 LRR_SD=0.1321 BAF_mean=0.5063 BAF_median=0.5000 BAF_SD=0.0391 BAF_DRIFT=0.000070 WF=-0.0113 GCWF=-0.0035
NOTICE: Reading LRR and BAF for chr1-22 from sample3.txt ... Done with 561288 records in 24 chromosomes (178 records are discarded due to lack of PFB information)
NOTICE: Adjusting LRR by GC model: WF changes from -0.0177 to 0.0135, GCWF changes from -0.0104 to 0.0047
NOTICE: Data from chromosome X,Y will not be used in analysis
NOTICE: Median-adjusting BAF values for all autosome markers from sample3.txt by 0.0268
NOTICE: quality summary for sample3.txt: LRR_mean=0.0024 LRR_median=0.0000 LRR_SD=0.1249 BAF_mean=0.5045 BAF_median=0.5000 BAF_SD=0.0431 BAF_DRIFT=0.000186 WF=0.0135 GCWF=0.0047
Compared to previous LOG messages, we can see that several extra lines are printed out, describing the changes in WF values and GCWF values. Basically WF measures the total signal fluctuation of a signal intensity file, while GCWF measures the GC-content-caused fluctuation of a signal intensity file. The signal adjustment procedure tries to reduce GC-content-caused fluctuation, and you will generally see a drop in the absolute value of GCWF after adjustment.